
Translate this page into:
Case Report on Rare Clinical Variant of Porokeratosis: Disseminated Superficial Porokeratosis
Address for correspondence: Dr. Jagdish J. Sakhiya, Sakhiya Skin Clinic, 2nd Floor, Ayush Doctor House, Station-Lal Darwaja Road, Surat 395003, Gujarat, India. E-mail: sakhiya.acedemic@rediffmail.com
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Wolters Kluwer - Medknow and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Abstract
Porokeratosis is a genodermatosis, resulting from a disorder in keratinization due to an abnormal clone of epidermal precursor, presenting with various clinical manifestations but characterized histologically by the presence of cornoid lamella. Diverse clinical variants of porokeratosis exist, which are unified by this general histological feature, but differ in morphology, distribution, and clinical course. The typical lesions of porokeratosis are described by an atrophic center surrounded by an elevated keratotic rim formed by the cornoid lamella. The lesions can be found almost anywhere on the body. We report an unusual presentation of a rare clinical variant, disseminated superficial porokeratosis.
Keywords
Cornoid lamella
disseminated superficial porokeratosis
porokeratosis
Disseminated superficial porokeratosis is the rare clinical variant of porokeratosis, which can be treated with acitretin resulting in cosmetic improvement. This case report opens the way for the future treatment option to deal with such a rare entity.

INTRODUCTION
Porokeratosis is a primary disorder of epidermal keratinization, described by annular plaques with an atrophic center and hyperkeratotic edges. Presence of cornoid lamella, a column of parakeratotic cells that occupies the small epidermal invaginations, is characteristic histopathological finding essential for the confirmation of diagnosis. It includes a group of heterogeneous disorders that represent various phenotypic expressions of the equivalent genetic defect, which is mainly inherited in an autosomal-dominant form. It can be classified into localized and disseminated forms.[1] We report an unusual presentation of a rare clinical variant of porokeratosis, disseminated superficial porokeratosis (DSP), which was treated with acitretin. The rarities of this variant and its clinical exuberance have motivated this report.
CASE REPORT
A 34-year-old brown skin man presented with multiple annular lesions, which firstly originated on upper limb and gradually extended to involve lower limb, chest, back, and thighs, bilaterally since last 2 years [Figure 1]. Dermatological examination revealed that initially, the lesions started appearing as papule with dimensions of 1–3 mm, brown in color, then slowly progressed in size of 8–10 mm as plaques with a keratotic ring. Lesions were asymptomatic and contained the pale, thinned central area with a ridgelike border, and yet some are red scaly and dry. Family members were also examined and no important cutaneous alterations were found. Routine laboratory investigations and X-ray chest were normal, and report of human immunodeficiency virus (HIV) was also unremarkable. The patient had no signs and symptoms of malignancy. History of radiation, drug intake, or heavy sun exposure was noncontributory. Specimen for a punch biopsy was taken from the lower limb. Histopathological examination revealed a keratin-filled epidermal invagination with a parakeratotic column, characteristic of cornoid lamella with sparse superficial perivascular lymphocytic infiltrate with focal interface change, and an absence of the granular layer below the cornoid lamella. The upper dermis and the stratum corneum have shown scattered clumps of necrotic keratinocytes with pigment dumplings. On the basis of the clinical and histopathological findings, the definite diagnosis of porokeratosis was made [Figure 2]. The patient was started with oral acitretin (25 mg oral daily) for 7 months. After treatment, a satisfactory improvement was observed over the upper limb, lower limb, chest, back, and thighs in the form of repigmentation [Figure 3]. The patient was continuing with the treatment at the time of writing this article. Currently, the patient is on the same treatment.
![Clinical variant of porokeratosis[1]](/content/173/2020/13/2/img/JCAS-13-145-g001.png)
- Clinical variant of porokeratosis[1]
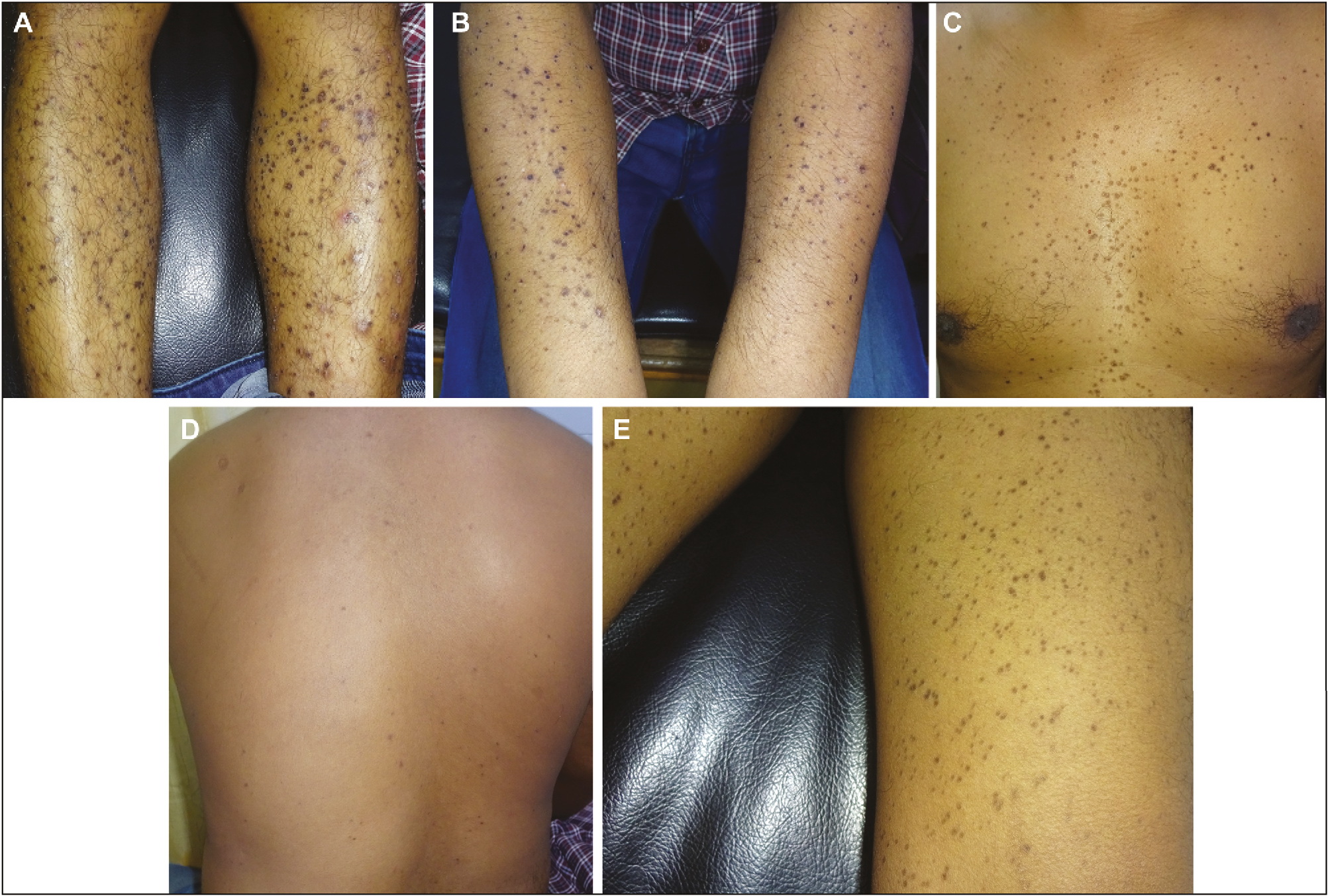
- Disseminated superficial porokeratosis: (A) lower limbs, (B) upper limbs, (C) chest, (D) back, (E) thighs
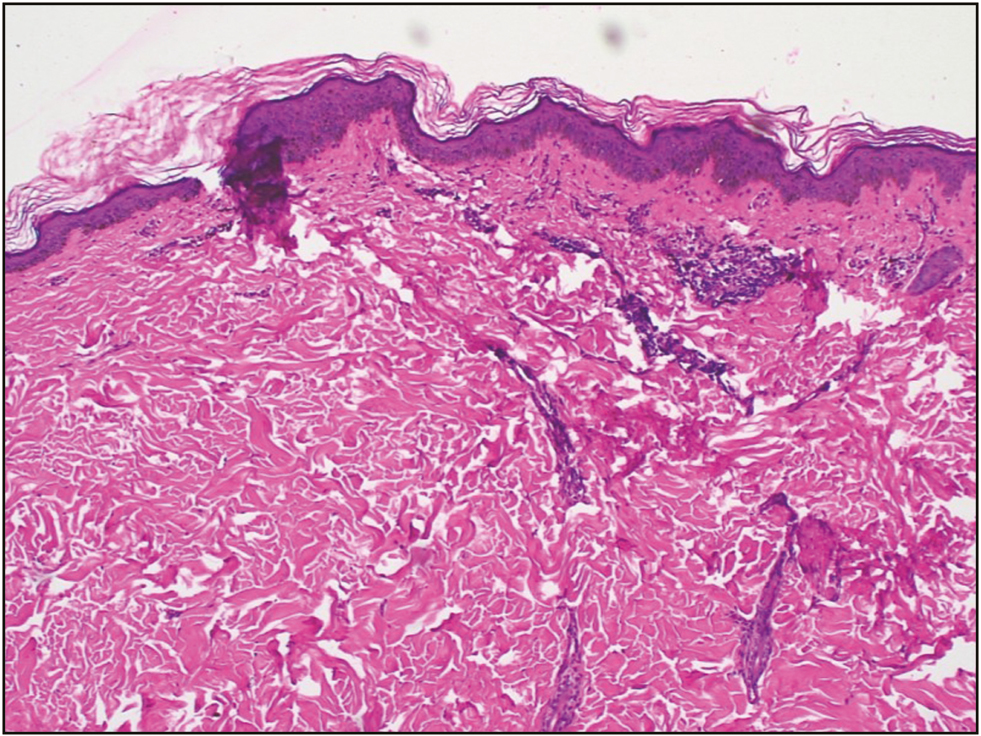
- Histopathological finding: cornoid lamella (hematoxylin and eosin, ×5)
DISCUSSION
In 1893, Vittorio Mibelli[2] used the term “porokeratosis” for the first time, which originated from the Greek word “poros” (pore of sweat gland) and “keratosis” (horny thickening). Porokeratosis manifests as scaly papules or plaques with a fine, elevated keratotic border. In Histopathological findings, presence of a column of porokeratosis, which is known as the cornoid lamella, overlies dyskeratotic keratinocytes and an absent granular layer was observed.
The six prominent clinical variants of porokeratosis are porokeratosis of Mibelli (PM), linear porokeratosis, punctate porokeratosis, DSP, disseminated superficial actinic porokeratosis (DSAP), and palmoplantar disseminated porokeratosis [Figure 4].[1] DSAP is the most common variant of porokeratosis. Its onset is usually in the third or fourth decade of life and involves the sun-exposed skin. It is often both induced and exacerbated by exposure to ultraviolet light, although the face is involved in only 15% of cases.[3] DSP, a rare variant, is distinguished from DSAP by its involvement of both sun-protected and sun-exposed areas of the body. DSP is characterized by small, annular, and brown lesions with raised hyperkeratotic borders and atrophic centers following the progression of the disease. Early lesions of DSP are small keratotic papules with central dell.[1] They may be erythematous or pigmented. They enlarge to form superficial ringlike lesions with slight central atrophy surrounded by discrete ridge topped by furrow. Females are more affected than males.[4] Furthermore, DSP tends to present in children or immunocompromised patients.[3] In our case, the patient had lesions in both sun-protected and sun-exposed areas of the body, and histological findings of the presence of cornoid lamellae have given an opinion in favor of DSP.
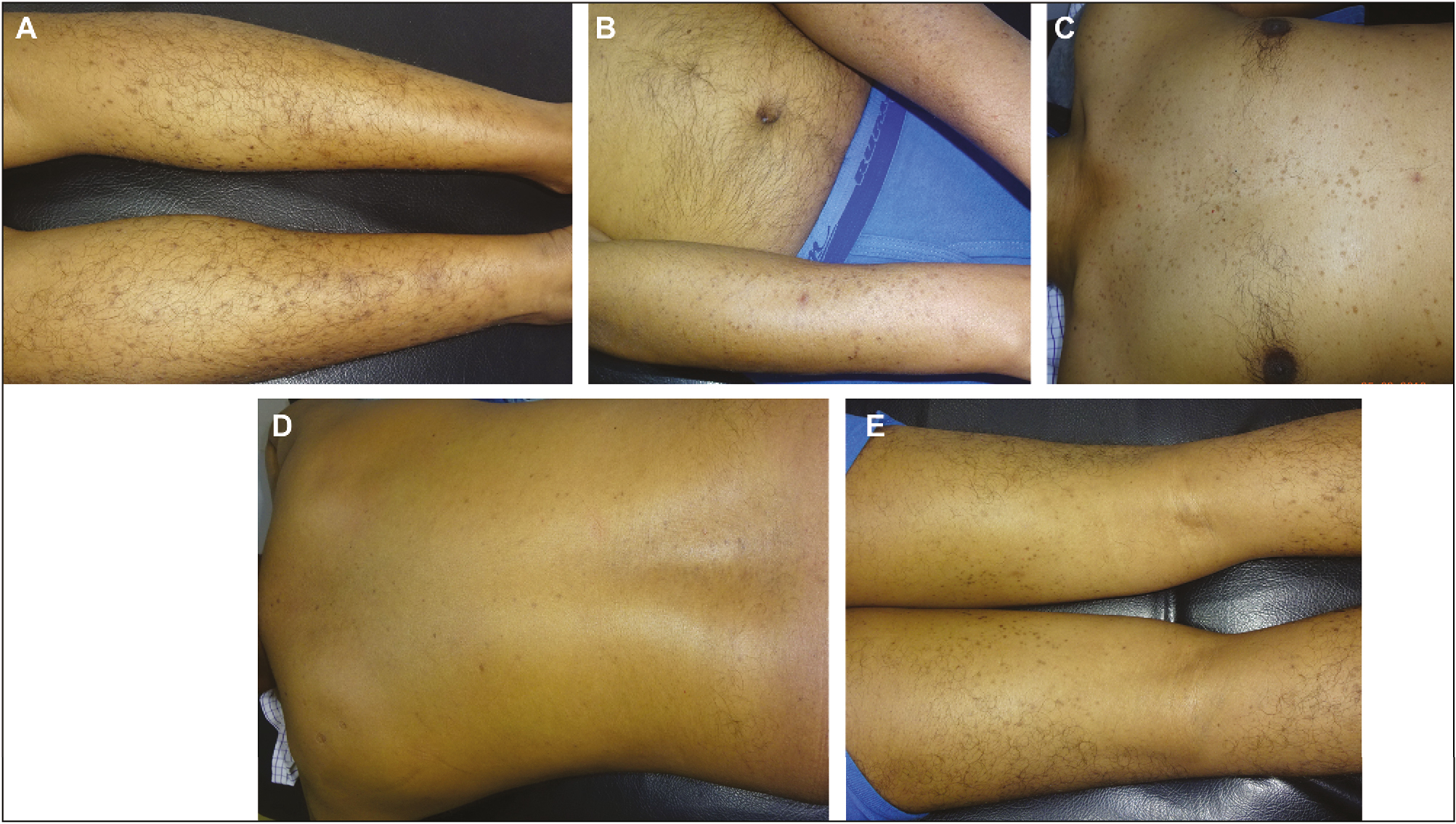
- Satisfactory improvement after acitretin treatment over (A) lower limbs (B) upper limbs (C) chest (D) back (E) thighs
DSP most commonly presents between the ages of 5 and 10 years but may develop later in life in an immunocompromised setting.[1] However, our patient had DSP in his third decade of life and had no immunocompromising conditions. It mainly involves extremities in a bilaterally symmetric manner. Lesions spare the axillary vaults, inguinal folds, perigenital region, palms, soles, and mucous membranes.[4] Our clinical photographs capture a rare presentation involving the upper limb, which gradually extended to involve lower limb, chest, back, and thighs bilaterally.
Porokeratosis is largely believed to arise from an expansion of abnormal keratinocytes, but the pathogenesis is not well delineated. In this theory, the cornoid lamella correlates with the border between the normal epidermis and the clone of mutant keratinocytes. Additional proposed etiologic or triggering factors include genetics, ultraviolet light, trauma, infection, and immunosuppression.[1] The genetic basis for DSP is unknown, but several genetic loci have been identified to date. Among these loci, a genetic locus for DSP has been found at 18p11.37.[5] Regarding the mode of inheritance, it may arise in an autosomal dominant form, more commonly in a random form. This was the presumable aetiology in our patient because no relatives were affected by a similar disease.[1] Nonetheless, a genetic evaluation for this patient and his family is a cornerstone to address genetic counseling issues for the future. Clinical distinct features of porokeratosis make it easy to diagnose, but, sometimes, the absence of the thready, keratotic ridge usually leads to other possible differential diagnoses including annular lichen planus, annular syphilide, annular sarcoidosis, and others.[6] In such confusing cases, a skin biopsy is carried out, and the specimen should be obtained from the peripheral hyperkeratotic ridge.
The therapeutic approach must be individualized, based on the size of the lesion and the anatomical location, the functional and aesthetic considerations, the risk of malignancy, and the patient’s preference. Protection from the sun, use of emollients, and watchful observation for signs of malignant degeneration are the general supportive stratagem recommended in many patients. In the case of wide-spreading of lesions, medical treatment is desired, and it gives potential benefit. Topical 5-fluorouracil is advisable in all variants of porokeratosis. Apart from this, topical vitamin D3 analogues (both calcipotriol and tacalcitol), immunomodulators (topical imiquimod cream and ingenol mebutate), and calcineurin inhibitors (tacrolimus 0.1%) have been shown to be useful in the treatment of DSAP, classic PM, and linear porokeratosis, respectively. Surgical treatment, which includes excision, cryotherapy, diamond fraise dermabrasion, also electrodesiccation, and curettage are essential for treating porokeratosis lesions that have undergone malignant transformation, and selection of treatment truly depends on the variant type. Various laser therapies are also available according to the variant type.[1] On the basis of our literature review, we found that the frequency-doubled neodymium-doped yttrium aluminum garnet; Nd:Y3Al5O12 (Nd:YAG) laser had a promising result in DSP yet restricted by the lack of evidence as only treated in one case report.[5] In summary, for DSP, only two treatments, including 5-Fluorouracil and frequency-doubled Nd:YAG were reported in the literature, yet they had a lack of strong evidence. As expected, systemic acitretin may be beneficial for DSP. With our empirical experience and deep knowledge, we have treated the patient with acitretin, which resulted in the improvement in cosmesis. This article may be helpful to other clinicians to treat such a rare entity in future.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgement
We are thankful to Dr. Trusha Gajjar, Dr. Piyush Darji, and Nimish Dudhatra for supporting this work.
REFERENCES
- Porokeratosis. 2018. Medscape. Available from: https://emedicine.medscape.com/article/1059123-overview.
- [Google Scholar]
- Contributo allo studio della ipercheratosidei canali sudoriferi. G Ital Mal Vener. 1893;28:313-55.
- [Google Scholar]
- Porokeratosis. In: Fitzpatrick TB, Eisen AZ, eds. Dermatology in general medicine (5th ed). New York: McGraw-Hill; 1999. p. :624-8.
- [Google Scholar]
- Genital porokeratosis: a series of 10 patients and review of the literature. Br J Dermatol. 2006;155:325-9.
- [Google Scholar]




